Frequently Asked Questions
General
We are located in the Plant Biology Laboratories Building:
612 Wilson Road, Rm S-18
East Lansing, MI 48824
It is easiest to find us by entering through the south entrance (pink arrow on the map below). Once inside, immediately turn left and head down into the basement. In the basement, turn left, pass through the door and we are a little way down the hall on the right (second image below).


The Genomics Core lab is open 8 AM to 5 PM Mondays through Fridays, excluding University Holidays and closures.
Please note that drop-in appointments are not accepted. Please email gtsf@msu.edu to get in touch with us or drop into our Zoom office hour (weekdays from 3 - 4 PM).
No, but samples should be dropped off during normal business hours, from 8:00 AM and 4:30 PM. This is to ensure your samples will be gathered up by Genomics Core staff and stored securely inside the lab. Detailed instructions are available on our Sample Drop-off & Shipping page.
Yes, we accept sample shipments. We recommend using FedEx or UPS; FedEx is preferred for international shipments. Overnight shipments have recently been experiencing delays so we recommend you ship your samples no later than Wednesday to reduce the possibility of your package sitting over a weekend. Address shipments to:
Shari Tjugum-Holland
612 Wilson Road, S-18 D
East Lansing, MI 48824
Further details regarding sample shipment are found on our Sample Drop-off & Shipping page.
The best way to contact us is by email at gtsf@msu.edu. This is a shared email account that is monitored by all Genomics Core staff. Often, we can respond within the same day, but sometimes it may take one to two business days.
You can also drop into our Zoom office hour without an appointment. Zoom office hours are held Monday - Friday from 3 - 4 PM.
For services that require your submission to be made through our LIMS, you will need to request a LIMS account. These services include AVITI, Oxford Nanopore, TapeStation, Fragment Analysis/GeneScan, and Qubit Flex. To request a LIMS account click "Request for a User ID" link on the LIMS Login webpage. Account requests are manually reviewed by Genomics Core staff. If you do not receive an approval after 24 hours, please contact us at gtsf@msu.edu. Please note that the request approval email may go to your SPAM or Junk folder, please check these folders if you are expecting an approval.
A LIMS account is not needed for Sanger Sequencing submissions. Please download the most up to date submission form from our Forms webpage and submit a completed hardcopy with your samples. A Genomcis Depot account will be created for you when you submit samples the first time. Genomics Depot is where you will retrieve your data.
Element Biosciences AVITI (1)
Element Biosciences AVITI24 (1)
10x Genomics Chromium X
10x Genomics CytAssist for Visium
Oxford Nanopore PromethION P2 Solo (3)
Applied Biosystems 3730xl 96-capillary DNA sequencer (Ending mid 2026)
Agilent 4200 TapeStation (2)
Covaris M220 Sonicator
DeNovix CellDrop
DeNovix QFX Fluorometer
Invitrogen Qubit 1.0
Invitrogen Qubit 2.0
Invitrogen Qubit Flex
Sage Science Pippin Prep
Yes, general information on equipment and services can be found on the Resource Information for Grant Applications webpage. If you would like a Letter of Support, please email your request to gtsf@msu.edu.
Quality Control & Sample Prep
No. The NanoDrop does not accurately quantify nucleic acids and often overestimates the concentration, sometimes by as much as 10-fold. A fluorometric method, such as Qubit, Biotium, or PicoGreen, should be used for quantification. These methods use dyes that bind to the specific nucleic acid being measured.
If you do not have a Qubit in your lab/department, you can make a reservation to use the one in the Common Use Equipment Room for a small fee per sample. If you have ≥72 samples, they can be submitted for our Qubit Flex High Throughput Quantification service.
No. The Genomics Core has two TapeStation instruments, which provide equivalent assays to the BioAnalyzer. Our TapeStation services are described on our TapeStation Sample Requirements webpage.
Not necessarily. For photosynthetic plant samples that are run on the TapeStation, plastid rRNAs confound the TapeStation algorithm that calculates the RINe score. RINe scores of about 4.0 or 5.0 may still represent largely intact, high-quality RNA. Questionable TapeStation results from photosynthetic plant samples should be sent to the Genomics Core (gtsf@msu.edu) for review.
Below are examples of high-quality total RNA with low RINe scores. Examples A and B are total RNA from tomato leaf tissue. Examples C and D are total RNA from rice leaf tissue.
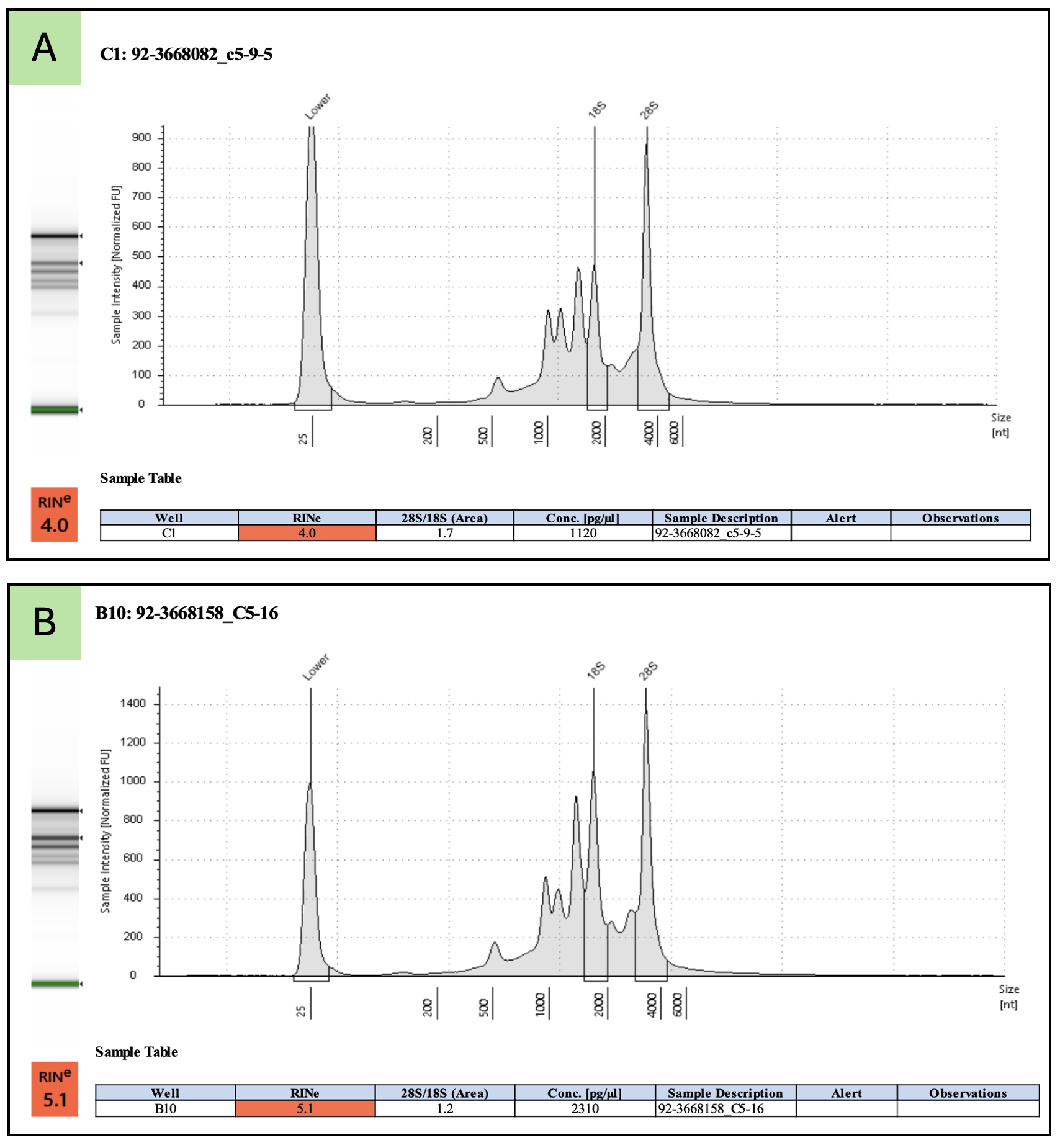
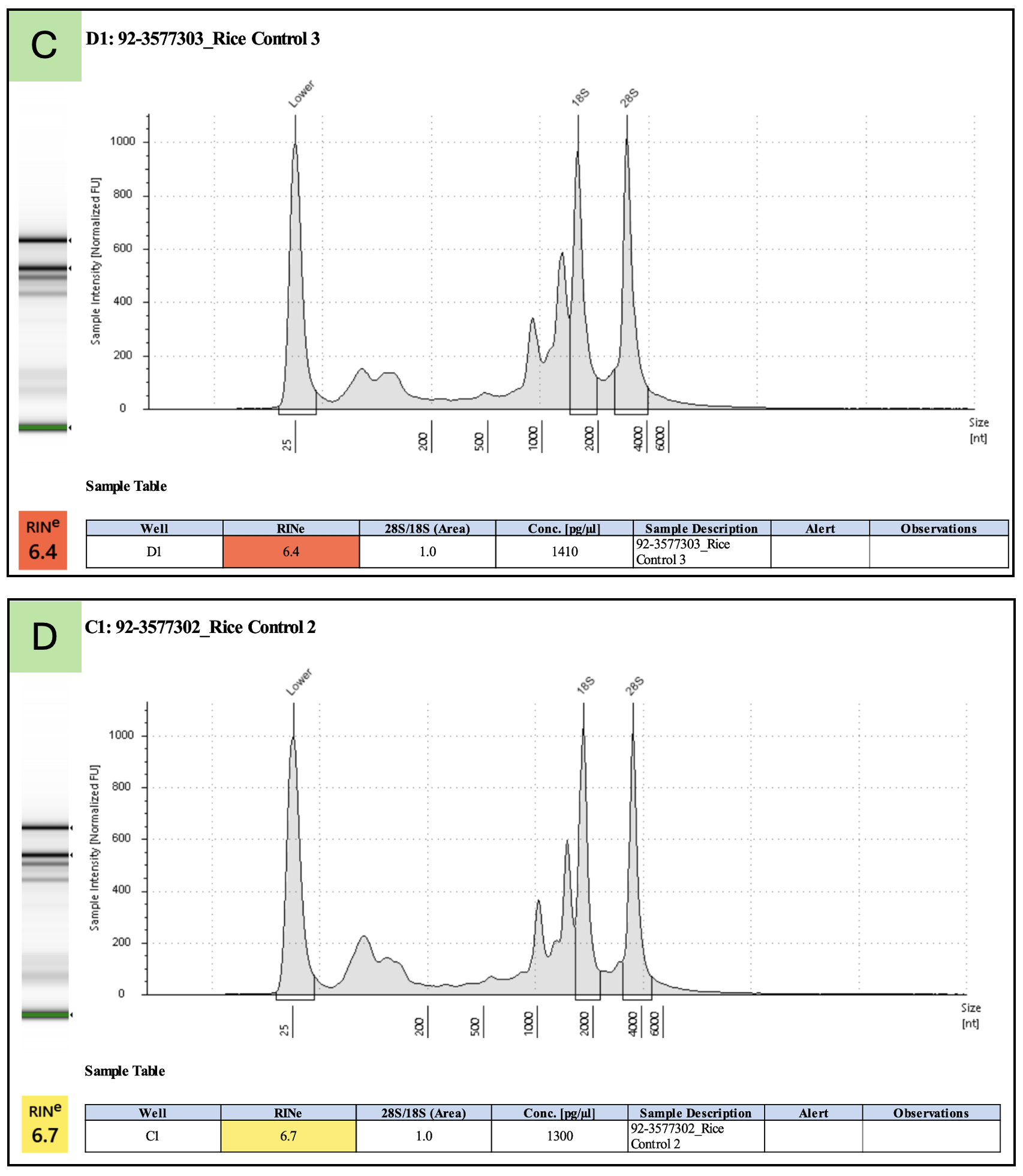
See this Agilent Application Note for a detailed description of the complications from plastid rRNA and more examples.
Total RNA samples that are submitted for library preparation and AVITI short-read sequencing should be DNA-free. The Genomics Core recommends the TURBO DNA-free kit (Invitrogen, catalog # AM1907) for DNase treatment.
DNA samples that are submitted for library preparation and AVITI short-read sequencing should be free of RNA.
Sample requirements are given on our Sample Requirements Webpage.
Yes, but there are caveats. In these cases, the Genomics Core cannot guarantee successful library preparation and/or quality of data. If your samples do not meet our sample requirements, a discussion is required prior to submission. Please contact us at gtsf@msu.edu.
The short answer is yes, this is a sample requirement for 16S V4 amplicon library preparation. With that being said, some researchers choose not to run test gels. If you choose not to run test gels and we cannot get your samples to amplify, we will assume that the issue lies with the sample, not with our processes. Some researchers choose to test a subset, but again, if an untested sample does not amplify, we will not go to lengths to get it to work.
The Genomics Core does not recommend a particular DNA extract kit. If you are able to extract DNA of high quality and quantity, then the kit should be fine.
Spin-column kits are not recommended for DNA that will be used for Oxford Nanopore long-read sequencing.
The Genomics Core does not recommend a particular RNA extract kit. If you are able to extract total RNA of high quality and quantity, then the kit should be fine. If a TRIzol extraction method is used, a spin/clean-up column should be used to ensure any residual TRIzol has been removed. Residual TRIzol can inhibit library preparation.
Total RNA that will be submitted for micro RNA or small RNA library preparation must be extracted with a kit that retains micro and small RNAs.
Project Planning
Yes. The Genomics Core has open office hours via Zoom, weekdays from 3 - 4 PM Eastern time. You may join the office hours with this Zoom link.
If this time is not convenient, or you would like to consult with multiple members of the Genomics Core you may schedule an individual consultation via Zoom. Contact us at gtsf@msu.edu to schedule a meeting time.
AVITI/Illumina (short-read) DNA/RNA libraries
For current Quantbio sparQ DNA and Watchmaker RNA libraries we have sufficient barcodes to multiplex 1,536 libraries.
Lexogen Small RNA, Lexogen QuantSeq 3' mRNA-Seq, and IDT xGen Methyl-Seq (formerly Swift) are limited to 96 barcoded libraries.
Short-read amplicon libraries
For sequencing of 16S-V4 or other amplicons on the Illumina MiSeq or AVITI instruments we can multiplex a maximum of 380 libraries per flow cell.
Oxford Nanopore DNA/RNA libraries
The Oxford Nanopore Native Barcoding Kit (DNA) and cDNA PCR Barcoding Kit (RNA) provide 24 barcodes.
The Oxford Nanopore Rapid Barcoding Kit (DNA) provide 96 barcodes.
Note: The Direct RNA Seqencing Kit now has barcoding options. Please contact the Genomics Core at gtsf@msu.edu to discuss your project.
For short-read sequencing on the AVITI, the Genomics Core has the ability to multiplex up to 1,536 samples for Watchmaker mRNA and Quantabio SparQ DNA libraries. The Genomics Core can multiplex a maximum of 384 samples Lexogen QuantSeq 3' and IDT xGen Methyl-Seq. A maximum of 96 Lexogen Small RNA-Seq libraries can be multiplexed. The Genomics Core can multiplex up to 384 samples for 16S-V4 amplicons and amplicon barcoding libraries, however four of the barcodes are reserved for internal negative controls; this leaves a maximum of 380 libraries in a single lane.
Researchers who prepare their own libraries may have different multiplexing capacities depending on the number of unique indexes they have.
Yes, we can sequence libraries using custom sequencing or index primers, with certain restrictions.
Projects submitted for sequencing on the AVITI must purchase whole flow cells. Custom primers on this instrument are applied to the entire flow cell.
Aliquots of the primer(s) must accompany each new submission. This is to avoid any possibility of confusing custom primer sets. When submitting custom primer(s) along with your libraries please label the tube(s) with the assigned LIMS project ID and primer type, for example 'CAR1111_Read1-primer'. At least 35 µl of custom primer(s) at a concentration of 100 µM must be provided per flow cell that will be sequenced.
Projects will be invoiced after completion. The Genomics Core processes internally billing at the end of each month. On-campus researchers do not receive invoices. The Genomics Core sends out invoices to off-campus researchers at the beginning of each month.
If you require expidited invoicing, please contact the Genomics Core at gtsf@msu.edu at the time of submission. The Genomics Core can provide an invoice when all samples for your project have been physically received in our lab. No invoices will be provided before samples have been submitted.
Project Creation & Submission Using LabLink
- AVITI sequencing
- Oxford Nanopore sequencing
- TapeStation DNA/RNA QC
- Qubit Flex high throughput DNA/RNA quantification
- GeneScan/Fragment Analysis
Submission for Sanger Sequencing and 10X do not currently use this portal for submission.
Project creation and sample submission using LabLink starts with completion of a Sample submission form. Submission forms are Microsoft Excel files; there are forms specific for each project type. Forms are available for download on our LIMS Project Submission page. You can also use the ‘LIMS Project Submission’ link in the right-hand menu on the Genomics Core web pages.
ALWAYS return to the LIMS Project Submission webpage to download a fresh copy of a submission form for each new project, do not re-use forms you have previously downloaded and saved. The Genomics Core regularly makes updates to these forms.
Detailed instructions for completing these forms are available in this document. A video walk-through of the process is posted on our LIMS Project Submission page.Proceed to the LIMS Login page and sign-in with your username and password. (If you don’t have a LabLink account yet see the question ‘How do I set up a LabLink LIMS account?’ above.) Click the Create icon (a blue plus) at the top, right-hand corner of the page. Fill in the fields with requested information. Fields marked with red asterisks are mandatory. As each page is completed proceed to the next by clicking the boxes at the bottom, right-hand corner.
Detailed, step-by-step instructions are available in this document, as well as in the video walk-through posted on our LIMS Project Submission page.
The Genomics Core has a Project Queue webpage. Projects submitted for AVITI and Nanopore sequencing are tracked on this page. Projects are organized by sequencing type, AVITI or Nanopore, selectable via the tabs below the table.
Projects are listed by their LIMS ID which is assigned when you create a project using your LabLink account. On this page you can quickly glance the current status of your project(s) and track their progress through their workflow.
Instructions for locating your LIMS ID are given here.
Short-Read (AVITI/Illumina) Sequencing Data & Data QC
Data is most commonly downloaded from this server using Secure FTP protocol (FTPS). A detailed tutorial on using FTPS to connect to and download your data may be found on our Data Retrieval webpage.
Alternatively, you may use Globus to transfer your data. Globus is designed for high speed transfer of large quantitites of data. Using Globus, you can transfer your data from the Genomics Core server to the MSU HPCC in a single step. MSU and many other research institutions provide Globus accounts for faculty, staff, and students.
If you would like to use Globus for data transfer, please contact the Genomics Core (gtsf@msu.edu) to set this up.
AVITI sequence data are provided as gzip compressed, FASTQ format files. FASTQ is the standard file format for storing sequence data along with quality scores. Gzip compressed FASTQ files end with the filename extension .fastq.gz.
MD5 checksums are calculated for all .fastq.gz files and stored in a file(s) alongside any .fastq.gz files in the same directory. These checksums may be used to validate the integrity your sequence files after downloading.
Data remains available for download on the Genomics Data Server for 60 days from date of availability. After that is retained in our archive storage for a period of time. The RTSF Genomics Core only guarantees retention of sequence data for six months from the date of availability. See our Data Retention Policy for more information.
Generally no, you will not need to demultiplex your data yourself. Sequence data for any libraries prepared by the RTSF Genomics Core will be demultiplexed by us. Likewise, if you have submitted libraries prepared yourself with Illumina compatible i5/i7 indexes the core will demultplex these as well using index ID information you provide at the time of submission.
There may be situations in which the core is unable to demultiplex data for you. If libraries include non-standard index formats, all sequence data will be returned as a single, large .fastq.gz file for single end read sequencing, or two files (R1 & R2) for paired end sequencing. Barcode sequences will be embedded within this data depending on the particulars of your library construction method. Contact the Genomics Core (gtsf@msu.edu) prior to submitting your custom libraries to ensure downstream demultiplexing will go smoothly.
We do not perform any trimming of sequence data prior to it being returned to researchers.
Even though it is not expected that these reads should contain any residual adapter sequences it is always best practice to use a primer trimming step in your data preprocessing to remove any anomalous reads. There are several popular trimming tools for you to choose from. Some examples are Trimmomatic , cutadapt, and bbduk (part of the BBMap toolkit). Each of these include either configuration files which contain the most common Illumina adapter sequences or documentation on how to specify them. Information about sequences to trim from Illumina compatible libraries is available from Illumina's adapter trimming bulletin.
The MSU Genomics Core amplifies the 16S-V4 region using dual indexed, Illumina compatible primers with 3’ ends complementary to the regions flanking V4. These primers are composed of the green “Illumina” portions and dark blue target specific portions of the construct shown above. When sequencing is performed on the MiSeq instrument custom Read 1 and Read 2 sequencing primers are used which are complementary to target specific sequences 515F and 806R respectively. The dashed red line indicates the span of the amplicon which must be covered by the two reads. For 16S-V4 this span is 253bp so each read covers nearly the entire region. Given this design, it is expected that the output reads will not include the 515F/806R sequences.

Even though it is not expected that these reads should contain any residual PCR primer sequences, it is always best practice to use a primer trimming step in your data preprocessing to remove any anomalous reads.
Primary PCR product from CS1 to CS2 inclusive is provided by the researcher. The MSU Genomics Core adds the Illumina compatible adapters. When sequencing is performed on the MiSeq instrument, custom Read 1 and Read 2 sequencing primers are used which are complementary to CS1 and CS2 respectively. The dashed red line indicates the span of the amplicon which must be covered by the two reads with sufficient overlap. For example, if 2x250bp paired end sequencing is performed and the length represented by the dashed line is 400bp then there will be 100bp of overlap between R1 and R2.

The MSU Genomics Core does not trim the TS-FWD or TS-REV sequences from the 5’ ends of your reads. Researchers should perform this trimming as part of their initial QC process prior to any secondary analysis.
The sequencing summary file (Excel) provides aggregate and per sample metrics for average Q-Score and percentage of base calls ≥ Q30.
The Genomics Core provides a FastQC report to accompany each .fastq.gz data file. A detailed description and tutorial for interpreting these reports is provided on our FastQC Tutorial & FAQ webpage.
Sanger
Data is typically returned to researchers within two business days. Expected turnaround time is given in the table below.
| Submission Day | Data Available By |
|---|---|
| Monday | Wednesday |
| Tuesday | Thursday |
| Wednesday | Friday |
| Thursday | Monday |
| Friday | Tuesday |
If your data is not available after two business days, please contact us at gtsf@msu.edu.
Sequence data is returned to researchers through Genomics Depot. Note that it is the researcher's responsibility to download and store their data. Genomics Depot is not for long-term storage and data is only guaranteed for 6 months. See our Data Retention Policy webpage for more information.
Sanger sequence data files are ab1 file format. Researchers will need software to view and analyze this file type. There are many software options available. Some options to explore are: Sequence Scanner, SnapGene, 4Peaks, Chromas Lite, Chromas Pro, GeneStudio Pro, UGENE, Chromaseq, Geneious, SeqMan Pro, Sequencer, Vector NTI Express, CodonCode Aligner, DNA Baser, BioLign, CAP3, ApE, and EveryVector. Please note that the software mentioned here may be free or be available for a fee. This is not an all-inclusive list.
There are many software options available. Some options to explore are: Sequence Scanner, SnapGene, 4Peaks, Chromas Lite, Chromas Pro, GeneStudio Pro, UGENE, Chromaseq, Geneious, SeqMan Pro, Sequencer, Vector NTI Express, CodonCode Aligner, DNA Baser, BioLign, CAP3, ApE, and EveryVector. Please note that the software mentioned here may be free or be available for a fee. This is not an all-inclusive list.